O que é a Síndrome de Wolf-Hirschhorn?
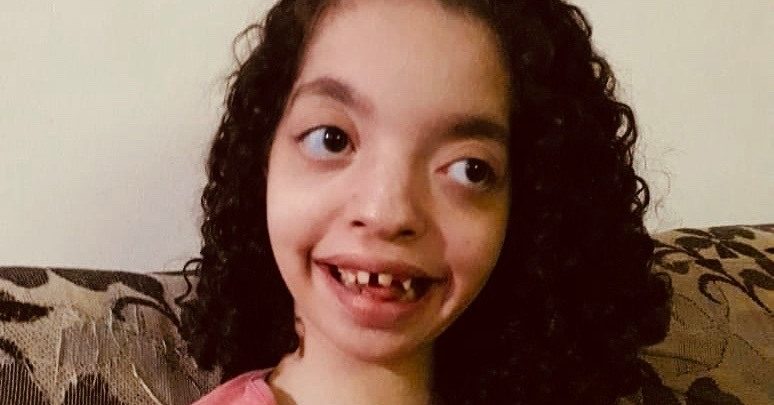

O que é a Síndrome de Wolf-Hirschhorn?
A síndrome de Wolf-Hirschhorn é uma condição genética rara que atinge o cromossomo 4. É um transtorno do desenvolvimento caracterizado por características craniofaciais típicas, deficiência de crescimento pré e pós-natal, atraso mental, atraso do desenvolvimento psicomotor grave, convulsões e hipotonia.
Causa
A Síndrome de Wolf-Hirschhorn é causada pela perda de unidades que compõem o material genético do braço curto do cromossomo 4, em que o paciente perde os genes NSD2, LETM1 e MSX1. Responsáveis respectivamente pela aparência facial distinta e o atraso no desenvolvimento, convulsões ou outra atividade elétrica anormal no cérebro e anormalidades dentárias e fenda labial.
Essa alteração cromossômica também é chamada de 4p-, e o tamanho da deleção varia entre os indivíduos afetados. Segundo a SWH Brasil, estima-se que a maioria dos casos (87%) trata-se de uma nova deleção, enquanto a outra parte (13%) é herdado de um dos seus progenitores como umas translocação cromossômica. Além disso, de 90 e 80% dos casos da síndrome de Wolf-Hirschhorn não são hereditários. Geralmente, a falha genética resulta de uma deleção cromossômica que ocorre como um evento aleatório durante a formação de células reprodutivas (óvulos ou espermatozóides) ou no desenvolvimento embrionário inicial.
Sinais e Sintomas
Todos os indivíduos com a Síndrome de Wolf- Hirschhorn apresentem deficiência de crescimento já durante o pré-natal e as principais características deste distúrbio incluem uma aparência facial característica, com ponte nasal ampla e plana, testa alta, microcefalia em 90% dos casos e o afastamento dos olhos e das órbitas oculares em excesso em 75%. Defeitos no couro cabeludo, sobrancelhas arqueadas, distância encurtada entre o nariz e o lábio superior, boca virada para baixo, queixo e orelhas pequenas também são outras características frequentes.
A condição interfere no crescimento, desenvolvimento e desencadeia deficiência intelectual (que pode variar entre grave e leve) e malformações cardíacas em 45% dos pacientes. Além disso, causa deficiência intelectual, convulsões e maior incidência no sexo feminino, que geralmente têm ausência do útero.
Alguns casos também incluem anomalias esqueléticas (60% -70%), perda auditiva (40%) e malformações do trato urinário.
Tratamento
O tratamento inclui reabilitação, terapia da fala, comunicação e linguagem de sinais. Ácido valpróico para convulsões e técnicas especiais de alimentação como gastrostomia. Tratamento padrão preventivo, como acompanhamento frequente para anomalias esqueléticas, oftalmológicas, defeitos cardíacos congênitos, perda auditiva, distúrbios do sono e adenomas hepáticos também são recomendados.
Além disso, é feita a prevenção de complicações secundárias com o uso de antibiótico para refluxo vesicoureteral, antibióticos contínuos para os casos com deficiências de anticorpos e acompanhamento sistemático para monitorar a reabilitação, hemograma completo anual, teste da função renal e ultrassonografia hepática de rotina.
Qual especialista procurar
Geneticista e pediatra
Estatísticas
Estima-se que a síndrome atinja 1 a cada 50.000 nascidos vivos. Todavia, essa estimativa pode não ser a estimativa real, pois, provavelmente muitos casos não sejam identificados.
Data comemorativa
O dia 16 de abril (16/04) foi escolhido como o Dia Internacional da síndrome porque a deleção acontece na região cromossômica 4p16.
O objetivo dessa data é promover a conscientização da existência dessa condição genética.
Deixe-nos saber o que você achou, porque sua opinião é muito importante para nós.
Foto: Arquivo pessoal/ Josi Santos